近日,云南大学与华大生命科学研究院联合在微生物领域国际权威期刊《微生物组》(Microbiome)(IF=12.6999)上发表了一项重要研究成果。该研究是我国重大标志性科学工程“第二次青藏高原综合科学考察研究”任务五“生物多样性保护和可持续利用”专题3“高原微生物多样性保护和可持续利用”的代表性科学任务之一,相关成果为今后推动我国青藏高原“动物微生物多样性保护和可持续利用”这一国家战略需求奠定了坚实的基础。研究团队基于大规模采集的高原代表性动物肠道微生物样本,开展了深度宏基因组测序分析,成功构建了迄今为止最完整的、规模最大的高原大型食草动物肠道菌群微生物组数据库,揭示了其中蕴含的丰富且尚未被充分开发的微生物资源。该资源库为开发新型基因编辑工具、抗菌肽等生物技术产品提供了重要支撑。同时,研究团队依托青藏高原这一独特自然实验室,在国际上首次证明了宿主与肠道菌群协同进化的新机制,为该领域的科研探索奠定了重要的理论基础。
构建迄今为止最完整的、规模最大的高原动物肠道微生物组数据库
研究团队历时五年,在青藏高原系统采集了5000余份大型食草动物的新鲜粪便样本。首期通过对其中1,412份样本(图1)进行宏基因组测序,团队获得了总量超过33TB的数据,并创新性地采用两种方法相互验证,成功构建起一个包含 14,062个高置信度的物种水平基因组箱(species-level genome bins, SGBs)的肠道微生物参考基因组集。
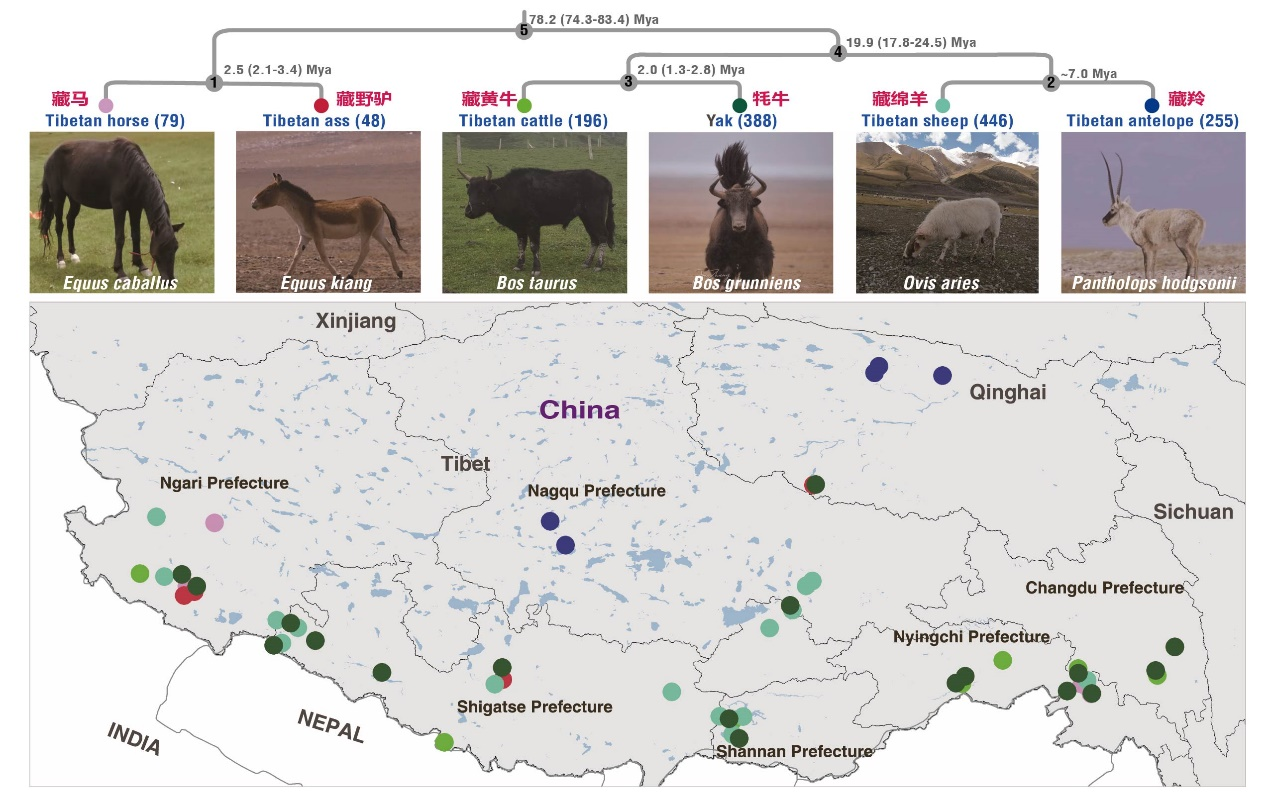
图1. 动物及采样分布图
值得一提的是,该数据库所包含的微生物物种中,有高达88%的物种为以往未被认知的新物种(图2)。这项研究首次在全基因组尺度上,大规模、系统地揭示了青藏高原特有大型食草动物肠道中蕴藏的宝贵微生物资源,标志着动物肠道微生物组研究领域进入了新的阶段。该研究整合了近期张志刚研究团队在Science China Life Sciences杂志联合公布的青藏高原六个哺乳动物目14个物种(涵盖人类、家畜和野生动物)的肠道微生物参考基因组集(包括了21,902个SGB,其中86%的SGB在现有数据库中未被注释)(文章链接:https://link.springer.com/article/10.1007/s11427-025-3047-5),研究团队完成了全球首个最大规模的青藏高原动物微生物资源库的一期工程建设(https://db.cngb.org/qtp/)。该数据库可以很好地服务于后续前沿科学问题的深入探索以及特色微生物资源的深度开发利用。
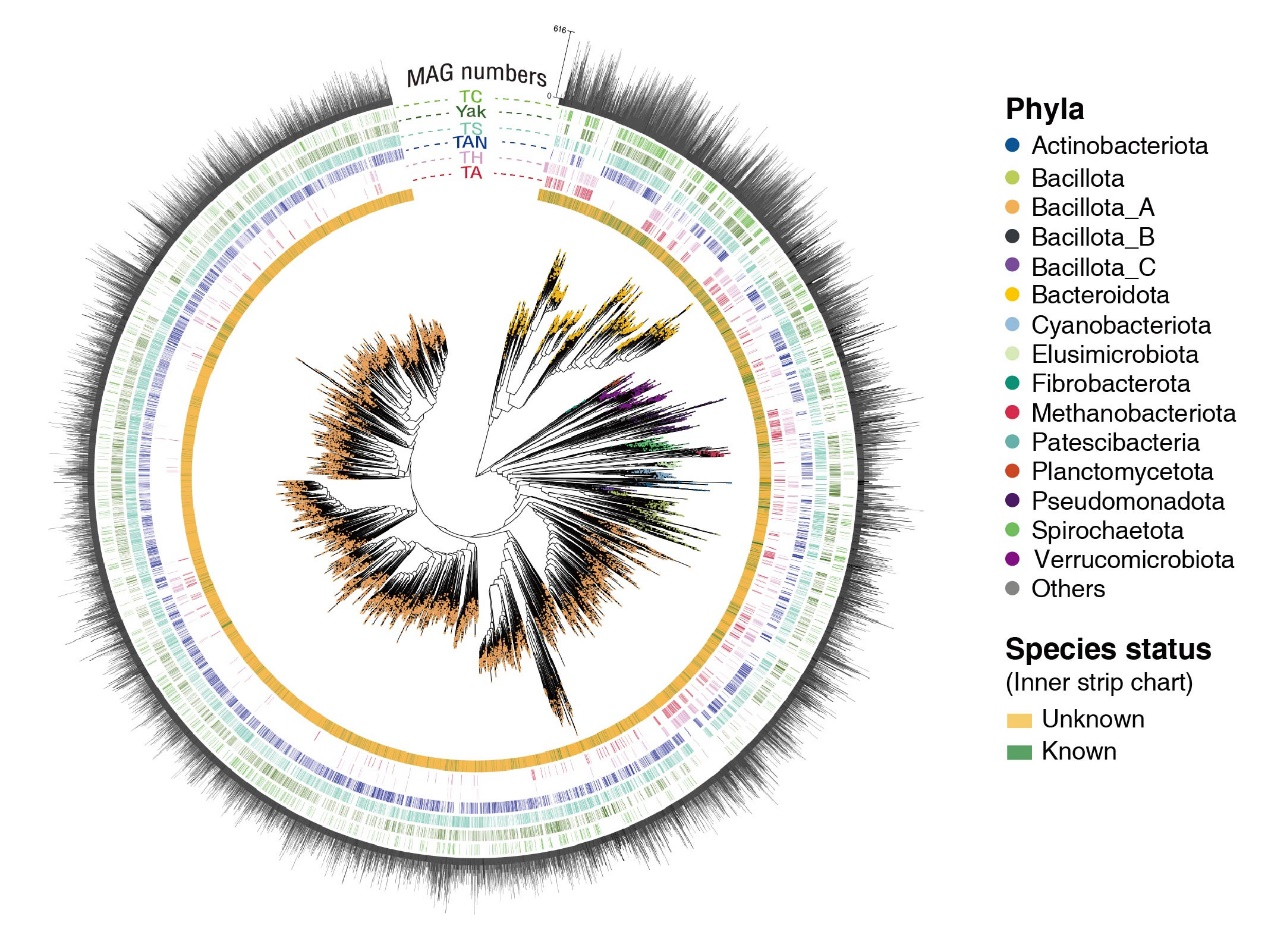
图2.14,062 个SGB的系统发育关系、分类注释以及宿主分布情况
通过对该基因组数据库进行深度挖掘,研究团队进一步发现,这些未知的肠道菌群蕴藏着数量庞大、潜力可观的生物合成基因簇(BGC)资源。研究证实,野生动物肠道菌群是一个仍被严重低估的微生物资源宝库,在生物技术和药物开发等领域具有巨大的探索价值。
项目启动初期,研究团队同步开展了纯培养工作,从青藏高原特色动物肠道中筛选出近3,000株单一菌株。其中,对牦牛来源的500多株菌进行的基因组分析表明,该地区动物肠道蕴含了大量未知的、多样性极高的微生物菌群(潜在新物种比例高达约38%)。同时,发现了其基因组中蕴藏着种类丰富且新颖性极高的次级代谢潜力。研究表明,超过94%的预测基因簇在现有数据库中无法找到同源物,预示着大量未知天然产物的存在。研究团队已从中发现多株功能性菌株,例如13株能高效降解纤维素的新型菌种,以及具有降低甲烷排放潜力的菌株,相关体外验证实验正在进行中。此项研究的初期成果已于今年在mSystems期刊上发表(文章链接:https://journals.asm.org/doi/10.1128/msystems.00367-25)。
解码“天然实验室”中的宿主-菌群共进化新模式
完整基因组集的构建,为研究肠道菌群与宿主的协同进化提供了强大支撑。本研究不仅深入揭示了非人类哺乳动物肠道微生物群的多样性、稳定性及功能特征,更重要的是,为阐明野生动物宿主与其共生肠道菌群的演化模式提供了重要的研究范式。
系统发育分析显示,沿宿主系统发育谱系反复发生的菌群获得与丢失事件,可能是驱动这些高原食草哺乳动物肠道微生物群组装的重要机制。对群体水平的分析进一步发现,在青藏高原生态系统中,宿主与其特定肠道共生微生物之间既存在频繁的共系统发育信号,也经常发生宿主交换事件(图3)。这标志着垂直传递和跨动物宿主之间的水平转移均为哺乳动物肠道微生物形成、维持和演化的主要驱动力,打破了之前“垂直传递”为主的学术观点。这不仅为理解动物宿主与肠道共生微生物的之间的演化关系提供了新的见解,也为今后有益微生物跨宿主应用奠定了重要的理论支撑。
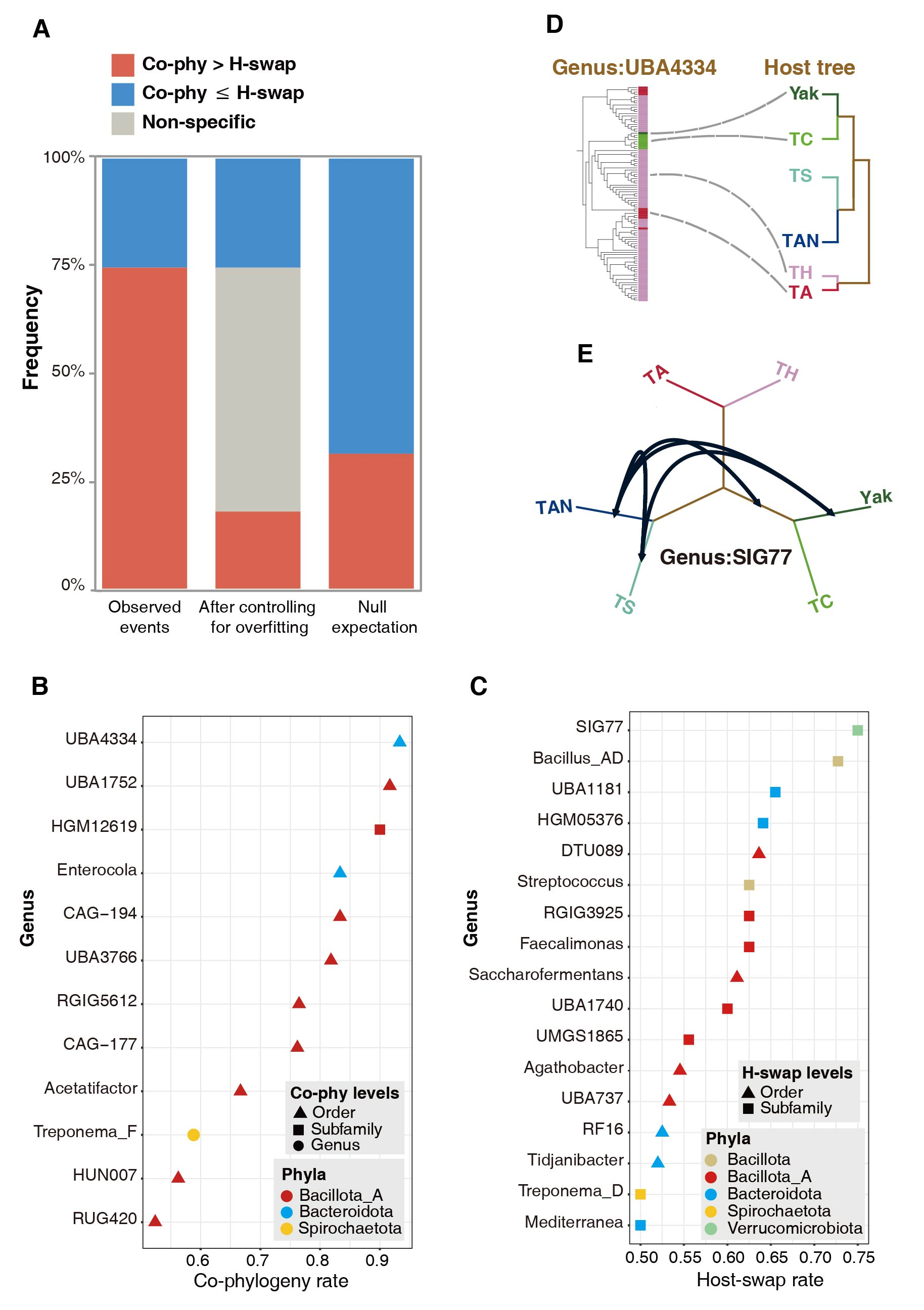
图3.共系统发生肠菌(D)与跨宿主交换肠菌(E)之间的频率比较(A-C)
武汉华大生命科学研究院生物技术副研究员李晓平、云南大学博士生田晨、博士毕业生庄道华和华大生命科学研究院硕士毕业生时兴伟为论文共同第一作者,云南大学研究员张志刚为论文通讯作者。
本研究得到西藏自治区科技计划项目(编号:XZ202401YD0012)、第二次青藏高原科学考察研究(STEP)计划(编号:2019QZKK0503)、云南省云岭学者(Z.Z.)、云南省地方科技发展中心指导资金(编号:202407AA110009)、国家自然科学基金(编号:U2002206 和 31970571)、云南省重大科技项目(编号:202001BB050001)以及云南大学研究生科研创新项目(编号:KC-22221159 和 ZC-22221199)的资助。
文章链接:
https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-025-02232-5
来源:国家重点实验室
编辑:奚利 责任编辑:李哲

